Preprocess
Requirement : length of first strand reads
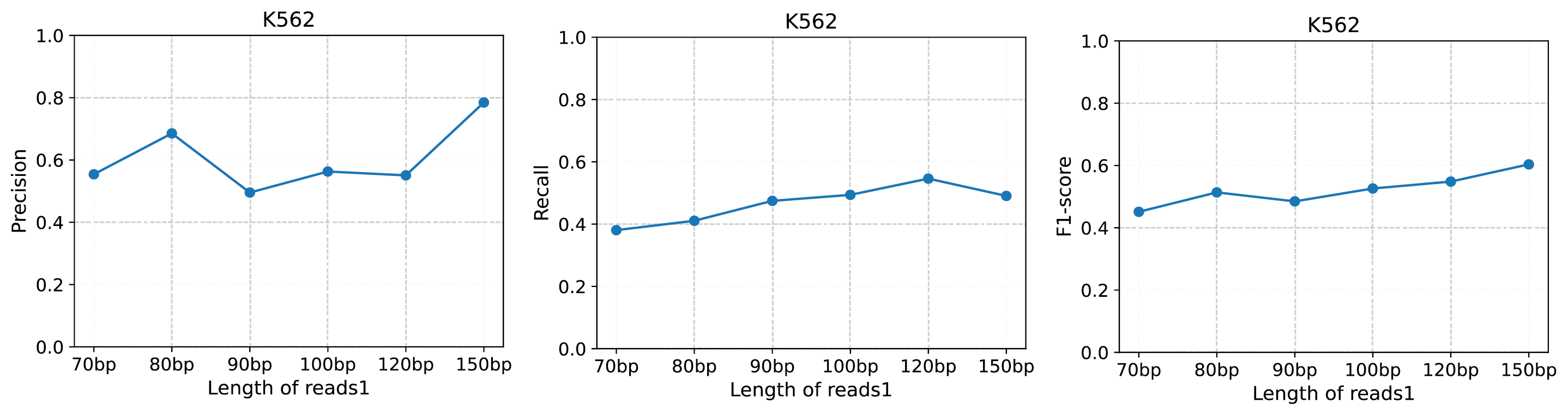
If you want to run scTail, you must make sure the length of your first strand reads is more than 70bp.
We evaluate different lengths of first strand read in K562 dataset and take the QuantSeq REV as the ground truth. The assessment result was shown in the following picture.
run STAR
scTail require 3’scRNA-seq data (10x Genomics) with the length of first strand reads more than 70bp. That means the first strand reads should contain extra cDNA information except UMI and cell barcode. You can follow below code to run STAR. You should change several parameters according to your data. If your 10x genomics data is V2 chemistry, then your cellbarcode length is 16bp, UMI length is 10bp,Poly(dT)VN length is 32bp. The –clip5pNbases should set as 58. –soloCBstart –soloCBlen –soloUMIstart and –soloUMIlen should set as 1, 16, 17 and 10, respectively. You also should download the cellbarcode whitelist such as 737K-august-2016.txt. For the 10x genomics V3 chemistry, the UMI length is 12bp. You should change other parameters according to this change. You also should change cellbarcode whitelist to 3M-february-2018.txt.
For more detail, you can refer to this website.
STAR --runThreadN 10 \
--genomeDir $human_STAR_index \
--readFilesIn reads_1.fastq.gz reads_2.fastq.gz \
--soloStrand Reverse \
--readFilesCommand zcat \
--soloType CB_UMI_Simple \
--soloBarcodeMate 1 \
--clip5pNbases 58 0 \
--soloCBstart 1 \
--soloCBlen 16 \
--soloUMIstart 17 \
--soloUMIlen 10 \
--outSAMtype BAM SortedByCoordinate \
--outFileNamePrefix $STAR_output_file_prefix \
--outFilterScoreMinOverLread 0.1 \
--outFilterMatchNminOverLread 0.1 \
--outFilterMultimapNmax 1 \
--outSAMattributes NH HI nM AS CR UR CB UB GX GN sS sQ sM \
--soloCBwhitelist 737K-august-2016.txt
Filter
scTail usually take alignment file (i.e. bam file) from STAR as input. Here, we try to filter out reads which do not contain GX, CB and UB.
Filter out reads according to the following code:
import pysam
inputbamfile=$home+'/STAR_output/SRR16796861Aligned.sortedByCoord.out.bam'
outputbamfile=$home+'/STAR_output/SRR16796861_GX_CB_UB_filtered.bam'
inputbam=pysam.Samfile(inputbamfile,'rb')
outputbam=pysam.Samfile(outputbamfile,'wb',template=inputbam)
for read in inputbam.fetch():
if (read.get_tag('GX')!='-') & (read.get_tag('CB')!='-') & (read.get_tag('UB')!='-'):
outputbam.write(read)
inputbam.close()
outputbam.close()
For large size samples
Sometimes, the bam file is too large (>30G). To run scTail smoothly and fastly, we should split
the large bam file to several samll bam files.
For the detail, you can check the manual of sinto.
sinto filterbarcodes -b big_sample.bam -c cellID_smallsample.tsv -p 20 --outdir $output_folder